Feature Review
Unveiling Cellular Heterogeneity in Colorectal Cancer Through Single-Cell Sequencing 
 Author
Author  Correspondence author
Correspondence author
International Journal of Molecular Medical Science, 2024, Vol. 14, No. 4
Received: 25 Jun., 2024 Accepted: 29 Jul., 2024 Published: 12 Aug., 2024
Intratumoral heterogeneity poses a significant challenge in the treatment and understanding of colorectal cancer. Recent advancements in single-cell sequencing technologies have provided unprecedented insights into the cellular diversity within colorectal tumors. This study explores the application of single-cell RNA sequencing (scRNA-seq) and multiomics approaches to dissect the complex heterogeneity of colon cancer at a single-cell resolution. By analyzing individual tumor cells, researchers have identified distinct subtypes of cancer-associated fibroblasts, cancer stem cells, and immune cell populations, each contributing uniquely to tumor progression and therapeutic resistance. The integration of transcriptomic, genomic, and epigenomic data has revealed the dynamic interplay between genetic mutations, epigenetic modifications, and gene expression patterns, offering a comprehensive understanding of tumor evolution and metastasis. Furthermore, computational tools and algorithms have been developed to enhance the clustering accuracy and interpret the high-dimensional data generated from single-cell analyses. These advancements underscore the potential of single-cell sequencing to inform personalized treatment strategies and improve clinical outcomes for colon cancer patients.
1 Introduction
Colorectal cancer (CRC) is one of the most common malignancies worldwide. It arises from the epithelial cells lining the colon or rectum and is characterized by a series of genetic and epigenetic alterations that drive the transformation of normal colonic epithelium into adenocarcinoma. The progression of CRC typically follows a well-defined sequence from benign adenomatous polyps to invasive cancer, often influenced by both hereditary and environmental factors (Punt et al., 2017; Bian et al., 2018). Advances in molecular biology have significantly enhanced our understanding of CRC, leading to the identification of key genetic mutations and pathways involved in its pathogenesis, such as the Wnt/β-catenin, TGF-β, and p53 signaling pathways (Chen et al., 2016; Punt et al., 2017). Cellular heterogeneity within tumors is a critical factor influencing cancer progression and prognosis. This heterogeneity arises from genetic, epigenetic, and phenotypic variations among cancer cells, leading to diverse subpopulations within a single tumor. Understanding this complexity is essential for developing effective therapeutic strategies. Single-cell sequencing technologies have revolutionized our ability to dissect this heterogeneity at an unprecedented resolution, providing insights into the distinct cellular populations and their roles in tumor biology.
Recent advancements in single-cell sequencing have enabled the simultaneous analysis of multiple omics layers, such as the genome, transcriptome, and epigenome, within individual cancer cells. For instance, Bian et al. (2018) utilized single-cell triple omics sequencing (scTrio-seq) to examine mutations, transcriptomes, and methylomes in colorectal cancer, revealing consistent DNA methylation patterns within genetic sublineages and substantial differences among clones. Similarly, single-cell RNA sequencing (scRNA-seq) has been employed to uncover transcriptional heterogeneity in various cancers, including glioblastoma and ovarian cancer, highlighting the presence of diverse cellular subpopulations with distinct gene expression profiles (Patel et al., 2014; Winterhoff et al., 2020).
The ability to analyze tumors at single-cell resolution has profound implications for precision medicine. By identifying specific genetic and phenotypic subclones, researchers can better understand the mechanisms driving tumor evolution, metastasis, and drug resistance. This knowledge can inform the development of targeted therapies and improve patient outcomes. For example, single-cell sequencing has been used to identify rare cancer stem cells and their role in metastasis and recurrence, providing potential targets for therapeutic intervention (Min et al., 2017).
High tumor heterogeneity of CRC poses significant challenges for effective treatment and prognosis. Single-cell sequencing allows for the detailed analysis of genetic and protein variations between individual cancer cells, offering a more comprehensive understanding of the tumor microenvironment and the mechanisms driving cancer progression and metastasis (Wen et al., 2023). This technology has also been applied to other cancers, such as liver cancer and high-grade serous ovarian cancer, revealing the complex interplay between cancer cells and their microenvironment (Tian and Li, 2022; Xu et al., 2022).
This study explores the application of single-cell sequencing in uncovering cellular heterogeneity in CRC. It summarizes advancements in single-cell sequencing technologies, highlighting their role in understanding intratumor heterogeneity and tumor evolution. It analyzes the tumor microenvironment (TME), discussing how single-cell sequencing characterizes the TME in CRC and identifies key cellular interactions and signaling pathways that contribute to cancer progression and treatment resistance. Additionally, this study reviews findings that identify novel biomarkers and therapeutic targets for more precise and effective CRC treatments. It also addresses current challenges and proposes future research directions to enhance the understanding of cellular heterogeneity in CRC.
2 Colorectal Cancer and Cellular Heterogeneity
2.1 Definition and significance of cellular heterogeneity
Cellular heterogeneity refers to the existence of diverse cell populations within a tumor, each with distinct genetic, epigenetic, and phenotypic profiles. This heterogeneity can be observed at multiple levels, including genetic mutations, gene expression patterns, and epigenetic modifications (Li et al., 2017; Bian et al., 2018; Min et al., 2020). In the context of colon cancer, cellular heterogeneity is a critical factor that contributes to tumor evolution, metastasis, and resistance to therapy. Single-cell sequencing technologies have been instrumental in uncovering the extent of this heterogeneity, revealing the presence of various subclones within tumors that may have different roles in cancer progression and response to treatment (Dalerba et al., 2011; Li et al., 2017; Min et al., 2020).
2.2 Impact of heterogeneity on cancer progression and treatment
The heterogeneity within colon cancer tumors has profound implications for disease progression and treatment outcomes. Different subclones within a tumor can exhibit varying degrees of aggressiveness, metastatic potential, and sensitivity to therapies. For instance, cancer stem cells (CSCs), which are a rare population within tumors, have been shown to drive metastasis and recurrence due to their ability to self-renew and differentiate into multiple cell types (Min et al., 2020). Additionally, the presence of distinct genetic and epigenetic profiles among tumor cells can lead to differential responses to chemotherapy and targeted therapies, making it challenging to achieve complete eradication of the tumor (Bian et al., 2018; Hamid et al., 2020; Xiang et al., 2021).
Single-cell sequencing studies have provided valuable insights into the mechanisms underlying tumor heterogeneity and its impact on treatment resistance. For example, the identification of specific genetic mutations and epigenetic modifications in single circulating tumor cells (CTCs) has highlighted the genetic diversity among these cells and their potential role in disease progression (Hamid et al., 2020). Furthermore, the development of prognostic gene signatures based on cell differentiation trajectories has shown promise in predicting patient outcomes and guiding personalized treatment strategies (Xiang et al., 2021).
In summary, understanding and addressing cellular heterogeneity in colon cancer is crucial for improving therapeutic strategies and achieving better clinical outcomes. The integration of single-cell sequencing technologies into clinical practice holds great potential for advancing precision medicine and tailoring treatments to the unique genetic and epigenetic landscape of each patient's tumor.
3 Single-Cell Sequencing Technologies
3.1 Introduction to single-cell sequencing
3.1.1 Historical perspective
The study of cellular heterogeneity in cancer has long been hindered by the limitations of bulk sequencing methods, which average out the signals from diverse cell populations, masking the complexity within tumors. The advent of single-cell sequencing technologies has revolutionized this field by enabling the analysis of individual cells, thus providing unprecedented insights into tumor biology. Early applications of single-cell sequencing focused on understanding the genomic and transcriptomic landscapes of individual cells, which has since expanded to include epigenomic and multi-omics approaches (Ren et al., 2018; Lei et al., 2021; Bowes et al., 2022). Figure 1 shows the workflow of single-cell sequencing.
|
Figure 1 New ICT based fertility management model in private dairy farm India as well as abroad |
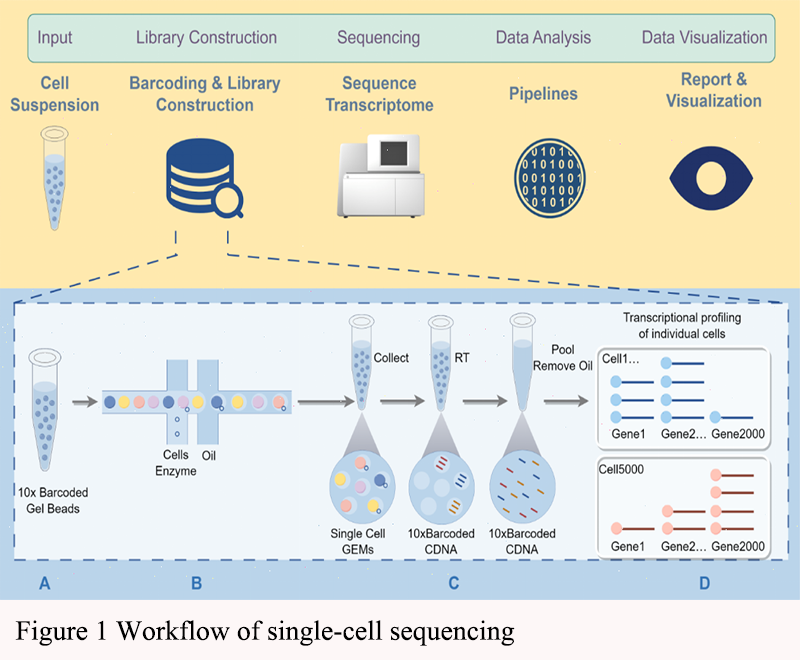
3.1.2 Technological advancements
Technological advancements in single-cell sequencing have been rapid and transformative. Initial methods were limited by low throughput and high costs, but recent innovations have significantly improved the efficiency, accuracy, and scalability of these techniques. For instance, the development of microfluidic platforms and droplet-based methods has enabled the high-throughput sequencing of thousands of individual cells simultaneously. Additionally, advancements in computational algorithms have enhanced the ability to analyze and interpret the complex data generated by single-cell sequencing, leading to more accurate clustering and characterization of cellular subpopulations (Ren et al., 2018; Ahmed et al., 2022; Bowes et al., 2022).
3.2 Types of single-cell sequencing
3.2.1 Single-cell RNA sequencing (scRNA-seq)
Single-cell RNA sequencing (scRNA-seq) is a powerful technique for profiling the transcriptomes of individual cells. This method has been instrumental in uncovering the cellular diversity within tumors and understanding the functional roles of different cell types in cancer progression. scRNA-seq has revealed distinct subpopulations of cancer cells and their interactions with the tumor microenvironment, providing insights into mechanisms of metastasis and therapy resistance (Li et al., 2017; Levitin et al., 2018; Ahmed et al., 2022). Recent advancements in scRNA-seq include the integration with spatial transcriptomics, which allows for the mapping of gene expression in the spatial context of tissue architecture (Ahmed et al., 2022).
3.2.2 Single-cell DNA sequencing (scDNA-seq)
Single-cell DNA sequencing (scDNA-seq) focuses on the genomic landscape of individual cells, providing detailed information on genetic mutations, copy number variations, and other genomic alterations. This technique has been pivotal in studying intratumor heterogeneity and tumor evolution, as it allows for the reconstruction of clonal architectures and the tracing of evolutionary dynamics within tumors. scDNA-seq has also been used to identify rare subclones that may contribute to disease progression and resistance to therapy (Navin et al., 2015; Bian et al., 2018; Bowes et al., 2022).
3.2.3 Single-Cell ATAC Sequencing (scATAC-seq)
Single-cell ATAC sequencing (scATAC-seq) is used to profile the chromatin accessibility landscape at the single-cell level. This technique provides insights into the regulatory elements and epigenetic modifications that govern gene expression in individual cells. scATAC-seq has been applied to study the epigenetic heterogeneity within tumors, revealing how different chromatin states can influence cellular behavior and contribute to cancer development. The integration of scATAC-seq with other single-cell omics techniques, such as scRNA-seq, has further enhanced our understanding of the complex regulatory networks in cancer (Bian et al., 2018; Ren et al., 2018; García-Sanz and Jiménez, 2021).
3.3 Technical considerations and challenges
Despite the significant advancements in single-cell sequencing technologies, several technical challenges remain. One major issue is the potential for technical noise and dropout events, where some transcripts or genomic regions may not be detected in every cell, leading to incomplete data. Additionally, the high cost and complexity of single-cell sequencing experiments can be limiting factors, particularly for large-scale studies. Another challenge is the need for robust computational tools to handle the vast amounts of data generated and to accurately interpret the results. Addressing these challenges will be crucial for the continued advancement and application of single-cell sequencing in cancer research (Ren et al., 2018; Lei et al., 2021; Ahmed et al., 2022; Bowes et al., 2022).
4 Application of Single-Cell Sequencing in CRC
Single-cell sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity in colon cancer. This technology allows for the dissection of complex tumor ecosystems at an unprecedented resolution, providing insights into the tumor microenvironment, identification of rare cell populations, clonal evolution, and the immune landscape.
4.1 Profiling tumor microenvironment
The tumor microenvironment (TME) plays a crucial role in the progression and treatment response of colon cancer. Single-cell sequencing has enabled detailed profiling of the TME, revealing the diverse cellular components and their interactions. For instance, scRNA-seq has been used to map the cell type-specific transcriptome landscape in various cancers, including lung cancer, which shares similarities with colon cancer in terms of TME complexity (Wu et al., 2021). This approach has identified rare cell types and highlighted the heterogeneity in cellular composition and intercellular signaling networks (Wu et al., 2021). Additionally, single-cell sequencing has provided insights into the immune microenvironment, showing how immune cells interact with tumor cells and influence disease progression (Chen et. al., 2023).
4.2 Identification of rare cell populations
One of the significant advantages of single-cell sequencing is its ability to identify rare cell populations within tumors. In advanced non-small cell lung cancer, scRNA-seq has uncovered rare cell types such as follicular dendritic cells and T helper 17 cells, which were not detected in previous studies using bulk sequencing methods (Wu et al., 2021). Similarly, in colon cancer, scRNA-seq can identify rare but potentially critical cell populations that contribute to tumor heterogeneity and treatment resistance. This capability is crucial for understanding the full spectrum of cellular diversity within tumors and for developing targeted therapies.
4.3 Understanding clonal evolution
Clonal evolution is a fundamental aspect of cancer biology, where tumor growth is driven by competing subclones. Single-cell sequencing has provided a powerful tool to study clonal evolution by tracking genetic and transcriptomic changes at the single-cell level. For example, in renal cell carcinoma, scRNA-seq combined with deep sequencing techniques has revealed the presence of subclonal single-nucleotide variants, supporting the clonal evolution model (Gerstung et al., 2012). This approach can be applied to colon cancer to understand how different subclones evolve, compete, and contribute to tumor progression and treatment resistance.
4.4 Mapping the immune landscape
The immune landscape of tumors is highly heterogeneous and plays a critical role in cancer progression and response to immunotherapy. Single-cell sequencing has been extensively used to map the immune landscape in various cancers. For instance, in breast cancer, scRNA-seq has profiled diverse immune cell phenotypes, revealing continuous phenotypic expansions specific to the tumor microenvironment (Azizi et al., 2018). In colon cancer, similar approaches can be used to map the immune landscape, identify key immune cell populations, and understand their roles in tumor immunity and immunotherapy response. This detailed mapping can inform the development of more effective immunotherapeutic strategies.
4.4.1 Differences immune microenvironment between left- and right- sided CRC
Right-sided CRC, originating from the cecum, ascending colon, and hepatic flexure, and left-sided CRC, originating from the splenic flexure, descending colon, and sigmoid colon, are often grouped as one disease. However, they represent clinically distinct entities with significant differences in prognosis and treatment outcomes (Gallois et al., 2018). Right-sided CRC typically has a worse prognosis than left-sided CRC (Stintzing et al., 2017). Extensive sequencing analyses have described a characteristic branching pattern of cancer evolution that supports the notion that tumor biology is characterized by both intratumor heterogeneity and the preservation of ancestral aberrations within the primary tumor and corresponding metastatic sites. The transcriptomes of 27 927 single human CRC cells from 3 left-sided and 3 right-sided CRC patients were analyzed by Guo et al. using single-cell RNA sequencing. This study revealed that right-sided CRC contains a significant proportion of exhausted CD8+ T cells with a highly migratory nature. In contrast, a cluster of cells from left-sided CRC exhibited states preceding exhaustion and a high ratio of preexhausted to exhausted T cells, which were identified as favorable prognostic markers. Additionally, these researchers identified a potentially novel subpopulation of cancer cells characterized by the expression of Retinol-binding protein 4 (RBP4) and Neurotensin (NTS), which exclusively expanded in left-sided CRC. Regulatory T cells (Tregs) from left-sided CRC showed higher levels of immunotherapy-related genes compared to those from right-sided CRC, suggesting that left-sided CRC may have increased responsiveness to immunotherapy. Furthermore, antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cellular cytotoxicity (ADCC) induced by M2-like macrophages were found to be more pronounced in left-sided CRC and correlated with a good prognosis in CRC. These findings provide valuable insights into the molecular and cellular differences between left-sided and right-sided CRC, which may help in the development of more targeted and effective treatment strategies for these distinct subtypes of CRC.
4.4.2 Differences in tumor microenvironment components between dMMR and pMMR CRC patients
Immunotherapy responses to cancer are highly variable, recent study demonstrated that tumors with deficient mismatch repair (dMMR) display a higher level of anti-tumor immunity when compared to tumors with proficient mismatch repair (pMMR) (Zhu et al., 2023). In order to better understand the potential mechanisms governing these diverse immune responses, Pelka et al. conducted a transcriptional profiling study with single-cell sequencing on 371 223 cells obtained from CRC and adjacent normal tissues of 28 individuals with pMMR and 34 individuals with dMMR (Pelka et al., 2021). Through an extensive analysis of 88 cell subsets and their associated gene expression programs, significant transcriptional and spatial remodeling was observed across the tumors. To identify key interactions between malignant and immune cells, the researchers identified expression programs that exhibited co-variation across tumors from affected individuals and utilized spatial profiling techniques to localize coordinated programs. Additionally, an immune hub enriched in dMMR tumors was discovered within the tumor microenvironment, characterized by the presence of activated T cells and the expression of chemokines that attract T cells, along with malignant and myeloid cells. By elucidating the interactions between different cellular programs, this study sheds light on the underlying logic that governs the spatial organization of immune-malignant cell networks.
4.4.3 Single cell sequencing reveals differences in immunotherapy efficacy ammong CRC patients
Although most of dMMR CRC patients received immune checkpoint inhibition immunotherapy could achieve good therapeutic efficacy, there are still a small number of patients suffered from unsatisfactory tumor regression. Li et al. employed single-cell RNA sequencing to examine the dynamics of immune and stromal cells in 19 dMMR CRC patients who received neoadjuvant PD-1 blockade (Li et al., 2023). This study revealed that in tumors achieving a pathological complete response (pCR), there is a coordinated decrease in CD8+ Trm-mitotic (tumor-resident memory T cells undergoing mitosis), CD4+ Tregs (regulatory T cells), proinflammatory IL1B+ Monocytes (monocytes expressing interleukin-1 beta), and CCL2+ Fibroblasts (fibroblasts expressing chemokine C-C motif ligand 2) following treatment. In contrast, the proportions of CD8+ Tem (effector memory T cells), CD4+ Th (helper T cells), CD20+ B (B cells expressing CD20), and HLA-DRA+ Endothelial cells (endothelial cells expressing human leukocyte antigen DRA) increased. They also discovered that proinflammatory features in the tumor microenvironment contribute to the persistence of residual tumors by modulating CD8+ T cells and other immune cell populations associated with treatment response. Chen et al. further explored the spatiotemporal cellular dynamics following neoadjuvant PD-1 blockade in CRC (Chen et al., 2024). They analyzed multiple sequential single-cell samples from 22 patients undergoing PD-1 blockade to map the evolution of local and systemic immunity in CRC patients. In this study, exhausted T (Tex) cells or tumor-reactive-like CD8+ T (Ttr-like) cells were found to be closely related to treatment efficacy. Additionally, Tex cells showed correlated proportion changes with multiple other tumor-enriched cell types following PD-1 blockade.
These findings provide valuable insights into the mechanisms underlying the response to PD-1 blockade in CRC patients. Understanding the relationship between these cell types and their changes in response to treatment may help in the development of more effective and personalized immunotherapy strategies for patients with CRC. Furthermore, these studies highlights the importance of single-cell analysis in unraveling the complex interactions between immune cells and tumor cells in the tumor microenvironment, which may ultimately lead to improved patient outcomes.
In conclusion, single-cell sequencing has provided profound insights into the cellular heterogeneity of colon cancer. By profiling the tumor microenvironment, identifying rare cell populations, understanding clonal evolution, and mapping the immune landscape, scRNA-seq has opened new avenues for research and therapeutic development in colon cancer.
5 Data Analysis and Interpretation
5.1 Preprocessing and quality control
Preprocessing and quality control are critical steps in single-cell sequencing to ensure the reliability and accuracy of the data. Initial steps involve filtering out low-quality cells and reads, which can be achieved by assessing metrics such as the number of detected genes per cell, the proportion of mitochondrial gene expression, and the overall read quality. For instance, Figure 1 in the study by Bian et al. (2018) scTrio-seq was employed to examine mutations, transcriptome, and methylome within colorectal cancer tumors, ensuring high-quality data by optimizing single-cell multiomics sequencing techniques. Similarly, quality control measures such as normalization and batch-effect correction are essential to mitigate technical variations and enhance the comparability of data across different samples (Li et al., 2021).
5.2 Data integration and dimensionality reduction
Data integration from multiple sources and dimensionality reduction are pivotal for managing the complexity of single-cell data. Techniques such as principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and uniform manifold approximation and projection (UMAP) are commonly used to reduce the dimensionality of the data while preserving its intrinsic structure. For example, in colorectal cancer research, integrating single-cell RNA sequencing (scRNA-seq) with bulk RNA transcriptome sequencing has revealed a heterogeneous immune landscape and pivotal cell subpopulations associated with prognosis (Zhang et al., 2023). These methods facilitate the visualization and interpretation of high-dimensional data, enabling the identification of distinct cellular populations and their relationships.
5.3 Clustering and cell type identification
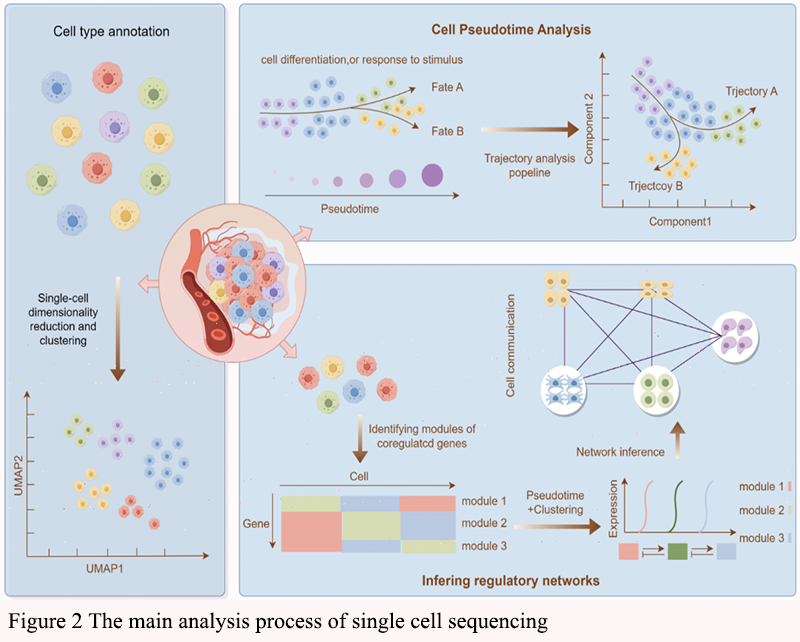
Clustering algorithms, such as k-means, hierarchical clustering, and graph-based methods, are employed to group cells with similar expression profiles. This step is crucial for identifying distinct cell types and subpopulations within the tumor microenvironment. In the study of advanced non-small cell lung cancer, single-cell RNA sequencing was used to map the cell type-specific transcriptome landscape, identifying rare cell types and revealing large heterogeneity in cellular composition and phenotype dominance (Figure 2) (Wu et al., 2021). Similarly, in colorectal cancer, single-cell sequencing has identified diverse genetic subclones within CD133 positive cancer stem cells, highlighting the complexity of tumor heterogeneity (Min et al., 2020).
|
Figure 2 The main analysis process of single cell sequencing |
.png){kind=link}
.png){kind=link}
5.4 Trajectory inference and lineage tracing
Trajectory inference and lineage tracing are advanced computational techniques used to reconstruct the developmental pathways and evolutionary trajectories of cells. These methods can elucidate the dynamic processes of cell differentiation and tumor progression. For instance, the study by Bian et al. (2018) demonstrated the feasibility of reconstructing genetic lineages and tracing their epigenomic and transcriptomic dynamics using single-cell multiomics sequencing. Additionally, trajectory models have been applied to understand the evolutionary trajectory from ulcerative colitis to colitis-associated cancer, providing insights into disease progression at the single-cell level (Wang et al., 2020). These approaches are instrumental in uncovering the mechanisms underlying cancer development and metastasis.
In summary, the integration of preprocessing, quality control, data integration, dimensionality reduction, clustering, cell type identification, trajectory inference, and lineage tracing in single-cell sequencing studies provides a comprehensive framework for unveiling cellular heterogeneity in colon cancer. These methodologies collectively enhance our understanding of tumor biology and hold promise for improving cancer diagnosis, prognosis, and treatment strategies. Figure 2 summarized the main analysis process of single cell sequencing.
6 Key Findings from Single-Cell Sequencing Studies
6.1 Case studies of single-cell analysis in CRC
Single-cell sequencing has been instrumental in uncovering the complexity of colon cancer at a granular level. For instance, a study on CRC utilized single-cell multiomics sequencing to analyze mutations, transcriptome, and methylome within tumors and metastases from patients. This approach provided insights into tumor evolution and linked DNA methylation to genetic lineages, revealing that DNA methylation levels are consistent within lineages but can differ substantially among clones (Bian et al., 2018). Another study focused on single circulating tumor cells (CTCs) from CRC patients, highlighting extensive genetic heterogeneities among CTCs and between primary tumors and CTCs. This genetic profiling suggested that single-cell genetic analysis could guide personalized therapeutic targets (Hamid et al., 2020).
6.2 Discovery of novel cell types and states
Single-cell RNA sequencing (scRNA-seq) has enabled the discovery of novel cell types and states within tumors. For example, a study on CD133 positive cancer stem cells in colorectal cancer identified heterogeneous subclones with distinct genetic variations, including specific mutations such as RNF144A and PAK2. This heterogeneity within cancer stem cells underscores the complexity of tumor biology and the potential for targeted therapies (Min et al., 2020). Additionally, scRNA-seq of colorectal cancer tissues revealed a diverse immune landscape, identifying 33 immune cell clusters and characterizing the heterogeneity of immune cell lineages in colon and rectal cancer (Zhang et al., 2023).
6.3 Insights into tumor-immune interactions
Single-cell sequencing has provided significant insights into tumor-immune interactions. A comprehensive CRC immune atlas restructured into 33 immune cell clusters revealed the heterogeneity of immune cell lineages and their interactions within the tumor microenvironment. The study identified pivotal cell subpopulations associated with colorectal cancer prognosis, such as CXCL13+ T cells and Ma1-SPP1 macrophages, which may promote angiogenesis and tumor progression (Zhang et al., 2023). Another study highlighted the role of immune cell heterogeneity in disease progression, emphasizing the importance of understanding immune cell interactions and regulatory roles in systems immunology and diseases (Chen et al., 2022).
6.4 Implications for personalized medicine
The findings from single-cell sequencing studies have profound implications for personalized medicine. By uncovering the genetic and epigenetic heterogeneity within tumors, these studies pave the way for more targeted and effective treatments. For instance, the identification of specific genetic subclones within cancer stem cells and CTCs can inform the development of personalized therapeutic strategies aimed at targeting these subpopulations (Hamid et al., 2020; Min et al., 2020). Moreover, the integration of single-cell and bulk RNA sequencing data to analyze immune cell heterogeneity and tumor microenvironment subtypes can help predict patient prognosis and tailor immunotherapy approaches (Zhang et al., 2023).
7 Challenges and Limitations
7.1 Technical and computational challenges
Single-cell sequencing technologies have revolutionized our understanding of cellular heterogeneity in colon cancer, but they come with significant technical and computational challenges. The isolation and sequencing of single cells require meticulous techniques such as laser-capture microdissection, fluorescence-activated cell sorting, and whole genome amplification, which are complex and resource-intensive (Schmidt and Efferth, 2016). Additionally, the high-dimensional data generated from single-cell RNA sequencing (scRNA-seq) necessitate advanced computational tools to extract meaningful biological insights. These tools must address issues such as distinguishing neoplastic from non-neoplastic cells, inferring cell communication within the tumor microenvironment, and delineating evolutionary trajectories of tumor cells (Fan et al., 2020). Moreover, the integration of data across multiple patients and disease states remains a formidable challenge, requiring robust algorithms to ensure accurate and reproducible results (Fan et al., 2020).
7.2 Biological interpretation of data
Interpreting the vast amount of data generated by single-cell sequencing poses another layer of complexity. The heterogeneity observed within tumors is not merely genetic but also phenotypic, involving diverse cellular programs that contribute to malignancy and treatment resistance (Muciño-Olmos et al., 2020). For instance, studies have shown that different cell clusters within a tumor can have distinct roles, such as promoting proliferation or enabling tissue invasion (Muciño-Olmos et al., 2020). This functional stratification complicates the biological interpretation of data, as researchers must decipher the specific contributions of each cell type to the overall tumor behavior. Additionally, the presence of cancer stem cells (CSCs) and their heterogeneous subclones further complicates the landscape, as these cells are known to drive metastasis and recurrence (Min et al., 2020). Understanding the evolutionary trajectory from normal tissue to cancerous states, as well as the role of specific genes in this progression, is crucial for developing targeted therapies (Wang et al., 2020).
7.3 Translational and clinical barriers
Despite the promise of single-cell sequencing in unveiling cellular heterogeneity, several translational and clinical barriers hinder its application in routine clinical practice. One major challenge is the need for standardized protocols and robust validation methods to ensure the reproducibility and reliability of single-cell analyses (Ren et al., 2018). Furthermore, the clinical utility of these technologies is limited by the current inability to efficiently translate single-cell data into actionable therapeutic strategies. For example, while single-cell sequencing can identify genetic subclones and potential therapeutic targets, integrating this information into personalized treatment plans remains a significant hurdle (Schmidt and Efferth, 2016). Additionally, the complexity of tumor ecosystems, including the interactions between cancer cells and the tumor microenvironment, adds another layer of difficulty in translating single-cell findings into clinical interventions (Ren et al., 2018). Finally, the high cost and technical demands of single-cell sequencing technologies pose practical barriers to their widespread adoption in clinical settings (Fan et al., 2020).
In summary, while single-cell sequencing offers unprecedented insights into the cellular heterogeneity of colon cancer, significant technical, computational, biological, and clinical challenges must be addressed to fully realize its potential in improving cancer diagnosis and treatment.
8 Future Directions and Emerging Technologies
8.1 Integrating single-cell and spatial omics
The integration of single-cell and spatial omics technologies represents a promising frontier in the study of colon cancer. Single-cell RNA sequencing (scRNA-seq) has already provided unprecedented insights into the transcriptomic landscape of individual cells within tumors, revealing the complexity of cellular interactions and heterogeneity (Ahmed et al., 2022). However, these techniques often lose spatial context, which is crucial for understanding the tumor microenvironment. Recent advancements in spatial transcriptomics and multiplexed imaging techniques allow for the detection of molecular biomarkers within their native spatial context, thereby offering a more comprehensive understanding of cell-to-cell variation within tumors (Lewis et al., 2021). This integration is expected to drive the next generation of research, improving diagnostic and therapeutic strategies by providing a holistic view of tumor biology.
8.2 Advances in single-cell multi-omics
Single-cell multi-omics technologies have revolutionized our understanding of tumor heterogeneity by enabling the simultaneous analysis of multiple molecular layers, such as genomics, transcriptomics, epigenomics, and proteomics, at single-cell resolution (Bian et al., 2018; Peng et al., 2020; Yu et al., 2023). Techniques like scTrio-seq and scONE-seq have demonstrated the feasibility of reconstructing genetic lineages and tracing their epigenomic and transcriptomic dynamics, providing insights into tumor evolution and metastasis (Bian et al., 2018; Yu et al., 2023). These advancements not only enhance our understanding of the molecular mechanisms driving cancer but also pave the way for the development of personalized therapies by revealing the complex interplay between different molecular layers.
8.3 Machine learning and AI in single-cell analysis
The application of machine learning (ML) and artificial intelligence (AI) in single-cell analysis is rapidly gaining traction. These technologies can handle the vast amounts of data generated by single-cell omics, identifying patterns and making predictions that would be impossible for humans to discern. For instance, ML algorithms can be used to infer cell-type-specific gene regulatory networks from scRNA-seq data, providing insights into the regulatory programs underlying cellular heterogeneity (Cha and Lee, 2020). Additionally, AI-driven approaches can integrate multi-omics data to uncover the complex interactions between genetic and non-genetic determinants of cancer evolution, thereby enhancing our understanding of tumor progression and resistance to therapy (Nam et al., 2020).
8.4 Prospects for clinical implementation
The ultimate goal of these technological advancements is their translation into clinical practice. Single-cell multi-omics has the potential to revolutionize cancer diagnosis and treatment by providing a detailed understanding of tumor heterogeneity and the molecular mechanisms driving cancer progression (García-Sanz and Jiménez, 2021; Pan and Jia, 2021). For instance, single-cell analyses can identify unique tumor clones and their spatial distribution within the tumor microenvironment, informing the design of targeted therapies (Tan et al., 2022; Yu et al., 2023). Moreover, the integration of single-cell and spatial omics can improve the accuracy of cancer diagnostics and prognostics, leading to more personalized and effective treatment strategies. As these technologies continue to evolve, their clinical implementation will likely become more feasible, offering new hope for cancer patients.
9 Concluding Remarks
The advent of single-cell sequencing technologies has significantly advanced our understanding of cellular heterogeneity in colon cancer. Studies have demonstrated that single-cell multiomics sequencing, such as scTrio-seq, can effectively reconstruct genetic lineages and trace epigenomic and transcriptomic dynamics within colorectal cancer tumors and metastases, providing insights into tumor evolution and the consistency of DNA methylation within genetic sublineages. Single-cell RNA sequencing (scRNA-seq) has revealed the diverse cellular populations within tumors, highlighting the transcriptional heterogeneity that can inform targeted combination therapies and clinical trial enrollment criteria. Additionally, single-cell sequencing has identified heterogeneous subclones within cancer stem cells, which are crucial for understanding metastasis and recurrence. The integration of high-sensitivity mutational analysis with RNA sequencing, as demonstrated by TARGET-seq, has further resolved the molecular signatures of genetically distinct subclones, offering insights into deregulated pathways in cancer cells.
The findings from single-cell sequencing studies underscore the importance of continuing to explore intratumoral heterogeneity to develop more effective cancer treatments. Future research should focus on improving the coverage and sensitivity of single-cell sequencing technologies to capture a more comprehensive picture of genetic and transcriptional variations within tumors. Additionally, longitudinal studies that track the dynamics of intra-tumor heterogeneity over time will be crucial for understanding how tumors evolve and adapt to therapeutic pressures. The identification of new transcriptional subpopulations and their role in metastasis highlights the need for further investigation into the mechanisms driving these changes and their potential as therapeutic targets. Moreover, integrating single-cell sequencing data with other omics data, such as proteomics and metabolomics, could provide a more holistic view of tumor biology and identify novel biomarkers for early detection and treatment response.
Single-cell sequencing has revolutionized our understanding of tumor heterogeneity in colon cancer, revealing the complex interplay between genetic, epigenetic, and transcriptional variations within tumors. These insights have significant implications for the development of personalized cancer therapies and the design of more effective treatment regimens. As the technology continues to advance, it holds the promise of uncovering new therapeutic targets and improving clinical outcomes for patients with colon cancer. The ongoing research in this field will undoubtedly contribute to a deeper understanding of cancer biology and pave the way for innovative approaches to cancer treatment.
Funding
This work was supported by Haiyan Foundation of Harbin Medical University Cancer Hospital (grant no.04000480).
Acknowledgments
The authors extend sincere thanks to two anonymous peer reviewers for their invaluable feedback on the manuscript of this paper.
Conflict of Interest Disclosure
The authors affirm that this research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahmed R., Zaman T., Chowdhury F., Mraiche F., Tariq M., Ahmad I., and Hasan A., 2022, Single-cell RNA sequencing with spatial transcriptomics of cancer tissues, International Journal of Molecular Sciences, 23(6): 3042.
https://doi.org/10.3390/ijms23063042
PMid:35328458 PMCid:PMC8955933
Azizi E., Carr A., Plitas G., Cornish A., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M., Choi K., Fromme R., Dao P., McKenney P., Wasti R., Kadaveru K., Mazutis L., Rudensky A., and Pe’er, D., 2018, Single-cell map of diverse immune phenotypes in the breast tumor microenvironment, Cell, 174: 1293-1308
https://doi.org/10.1016/j.cell.2018.05.060
Bian S., Hou Y., Zhou X., Li X., Yong J., Wang Y., Wang W., Yan J., Hu B., Guo H., Wang J., Gao S., Mao Y., Dong J., Zhu P., Xiu D., Yan L., Wen L., Qiao J., Tang F., and Fu W., 2018, Single-cell multiomics sequencing and analyses of human colorectal cancer, Science, 362: 1060-1063.
https://doi.org/10.1126/science.aao3791
Bowes A., Tarabichi M., Pillay N., and Loo P., 2022. Leveraging single‐cell sequencing to unravel intratumour heterogeneity and tumour evolution in human cancers, The Journal of Pathology, 257: 466-478.
https://doi.org/10.1002/path.5914
Cha J., and Lee I., 2020, Single-cell network biology for resolving cellular heterogeneity in human diseases, Experimental and Molecular Medicine, 52: 1798-1808.
https://doi.org/10.1038/s12276-020-00528-0
Chen D., Luo Y., and Cheng G., 2022, Single cell and immunity: better understanding immune cell heterogeneities with single cell sequencing, Clinical and Translational Medicine, 13(1): e1159.
https://doi.org/10.1002/ctm2.1159
Chen J., Zhou Q., Wang Y., and Ning K., 2016, Single-cell SNP analyses and interpretations based on RNA-Seq data for colon cancer research, Scientific Reports, 6(1): 34420.
https://doi.org/10.1038/srep34420
Chen S., Zhou Z., Li Y., Du Y., and Chen G., 2023, Application of single-cell sequencing to the research of tumor microenvironment, Frontiers in Immunology, 14: 1285540.
https://doi.org/10.3389/fimmu.2023.1285540
PMid:37965341 PMCid:PMC10641410
Chen Y., Wang D., Li Y., Qi L., Si W., Bo Y., Chen X., Ye Z., Fan H., Liu B., Liu C., Zhang L., Zhang X., Li Z., Zhu L., Wu A., Zhang Z., 2024, Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer, Cancer Cell, 2(7):1268-1285.e7.
https://doi.org/doi: 10.1016/j.ccell.2024.06.009
Dalerba P., Kalisky T., Sahoo D., Rajendra P., Rothenberg M., Leyrat A., Sim S., Okamoto J., Johnston D., Qian D., Zabala M., Bueno J., Neff N., Wang J., Shelton A., Visser B., Hisamori S., Shimono Y., Wetering M., Clevers H., Clarke M., and Quake S., 2011. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nature biotechnology, 29: 1120-1127.
https://doi.org/10.1038/nbt.2038
PMid:22081019 PMCid:PMC3237928
Fan J., Slowikowski K., and Zhang F., 2020, Single-cell transcriptomics in cancer: computational challenges and opportunities, Experimental & Molecular Medicine, 52: 1452-1465.
https://doi.org/10.1038/s12276-020-0422-0
Gallois C., Pernot S., Zaanan A., Taieb J., 2018, Colorectal cancer: why does side matter? Drugs, 78(8): 789-798.
https://doi.org/10.1007/s40265-018-0921-7
PMid:29790124
García-Sanz R., and Jiménez C., 2021, Time to move to the single-cell level: applications of single-cell multi-omics to hematological malignancies and Waldenström’s macroglobulinemia—a particularly heterogeneous lymphoma, Cancers, 13(7): 1541.
https://doi.org/10.3390/cancers13071541
Gerstung M., Beisel C., Rechsteiner M., Wild P., Schraml P., Moch H., and Beerenwinkel N., 2012, Reliable detection of subclonal single-nucleotide variants in tumour cell populations, Nature Communications, 3(1): 811.
https://doi.org/10.1038/ncomms1814
Hamid F., Gopalan V., Matos M., Lu C., and Lam A., 2020, Genetic Heterogeneity of single circulating tumour cells in colorectal carcinoma, International Journal of Molecular Sciences, 21(20): 7766.
https://doi.org/10.3390/ijms21207766
Lei Y., Tang R., Xu J., Wang W., Zhang B., Liu J., Yu X., and Shi S., 2021, Applications of single-cell sequencing in cancer research: progress and perspectives, Journal of Hematology & Oncology, 14(1): 91.
https://doi.org/10.1186/s13045-021-01105-2
Levitin H., Yuan J., and Sims P., 2018, Single-cell transcriptomic analysis of tumor heterogeneity, Trends in Cancer, 4(4): 264-268.
https://doi.org/10.1016/j.trecan.2018.02.003
Lewis S., Asselin-Labat M., Nguyen Q., Berthelet J., Tan X., Wimmer V., Merino D., Rogers K., and Naik S., 2021, Spatial omics and multiplexed imaging to explore cancer biology, Nature Methods, 18: 997-1012.
https://doi.org/10.1038/s41592-021-01203-6
Li H., Courtois E., Sengupta D., Tan Y., Chen K., Goh J., Kong S., Chua C., Hon L., Tan W., Wong M., Choi P., Wee L., Hillmer A., Tan I., Robson P., and Prabhakar S., 2017, Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors, Nature Genetics, 49: 708-718.
https://doi.org/10.1038/ng.3818
Li J., Wu C., Hu H., Qin G., Wu X., Bai F., Zhang J., Cai Y., Huang Y., Wang C., Yang J., Luan Y., Jiang Z., Ling J., Wu Z., Chen Y., Xie Z., Deng Y., 2023, Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer, Cancer Cell, 41(6):1152-1169.
https://doi.org/10.1016/j.ccell.2023.04.011
Li L., Xiong F., Wang Y., Zhang S., Gong Z., Li X., He Y., Shi L., Wang F., Liao Q., Xiang B., Zhou M., Li X., Li Y., Li G., Zeng Z., Xiong W., and Guo C., 2021, What are the applications of single-cell RNA sequencing in cancer research: a systematic review, Journal of Experimental and Clinical Cancer Research, 40(1): 163.
https://doi.org/10.1186/s13046-021-01955-1
Min D., Kim H., Kim J., Wen X., Kim S., Cho Y., Lim Y., Song S., Han S., Kwon S., Kang G., and Kim T., 2020, Phenotype-based single cell sequencing identifies diverse genetic subclones in CD133 positive cancer stem cells, Biochemical and Biophysical Research Communications, 558: 209-215.
https://doi.org/10.1016/j.bbrc.2020.09.005
Muciño-Olmos E., Vázquez-Jiménez A., León U., Matadamas-Guzmán M., Maldonado V., López-Santaella T., Hernández-Hernández A., and Reséndis-Antonio O., 2020, Unveiling functional heterogeneity in breast cancer multicellular tumor spheroids through single-cell RNA-seq, Scientific Reports, 10(1): 12728.
https://doi.org/10.1038/s41598-020-69026-7
Nam A., Chaligné R., and Landau D., 2020, Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics, Nature Reviews Genetics, 22: 3-18.
https://doi.org/10.1038/s41576-020-0265-5
Navin N., 2015, Delineating cancer evolution with single-cell sequencing, Science Translational Medicine, 7: 296fs29.
https://doi.org/10.1126/scitranslmed.aac8319
Pan D., and Jia D., 2021, Application of single-cell multi-omics in dissecting cancer cell plasticity and tumor heterogeneity, Frontiers in Molecular Biosciences, 8: 757024.
https://doi.org/10.3389/fmolb.2021.757024
PMid:34722635 PMCid:PMC8554142
Patel A., Tirosh I., Trombetta J., Shalek A., Gillespie S., Wakimoto H., Cahill D., Nahed B., Curry W., Martuza R., Louis D., Rozenblatt-Rosen O., Suvà M., Regev A., and Bernstein B., 2014, Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma, Science, 344: 1396-1401.
https://doi.org/10.1126/science.1254257
Pelka K., Hofree M., Chen J.H., Sarkizova S., Pirl J.D., Jorgji V., Bejnood A., Dionne D., Ge W.H., Xu KH., Chao S.X., Zollinger D.R., Lieb D.J., Reeves J.W., Fuhrman C.A., Hoang M.L., Delorey T., Nguyen L.T., Waldman J., Klapholz M., Wakiro I., Cohen O., Albers J., Smillie C.S., Cuoco M.S., Wu J., Su M.J., Yeung J., Vijaykumar B., Magnuson A.M., Asinovski N., Moll T., Goder-Reiser M.N., Applebaum A.S., Brais L.K., DelloStritto L.K., Denning S.L., Phillips S.T., Hill E.K., Meehan J.K., Frederick D.T., Sharova T., Kanodia A., Todres E.Z., Jané-Valbuena J., Biton M., Izar B., Lambden C.D., Clancy T.E., Bleday R., Melnitchouk N., Irani J, Kunitake H., Berger D.L., Srivastava A., Hornick J.L., Ogino S., Rotem A., Vigneau S., Johnson B.E., Corcoran R.B., Sharpe A.H., Kuchroo V.K., Ng K., Giannakis M., Nieman L.T., Boland G.M., Aguirre A.J., Anderson A.C., Rozenblatt-Rosen O., Regev A., Hacohen N., 2021, Spatially organized multicellular immune hubs in human colorectal cancer, Cell, 184(18): 4734-4752.
https://doi.org/10.1016/j.cell.2021.08.003
PMid:34450029 PMCid:PMC8772395
Peng A., Mao X., Zhong J., Fan S., and Hu Y., 2020, Single-cell multi-omics and its prospective application in cancer biology, Proteomics, 20(13): 1900271.
https://doi.org/10.1002/pmic.201900271
Punt C., Koopman M., and Vermeulen L., 2017, From tumour heterogeneity to advances in precision treatment of colorectal cancer, Nature Reviews Clinical Oncology, 14: 235-246.
https://doi.org/10.1038/nrclinonc.2016.171
Ren X., Kang B., and Zhang Z., 2018, Understanding tumor ecosystems by single-cell sequencing: promises and limitations, Genome Biology, 19(1): 211.
https://doi.org/10.1186/s13059-018-1593-z
Schmidt F., and Efferth T., 2016, Tumor heterogeneity, single-cell sequencing, and drug resistance, Pharmaceuticals, 9(2): 33.
https://doi.org/10.3390/ph9020033
Stintzing S., Tejpar S., Gibbs P., Thiebach L., Lenz H.J., 2017, Understanding the role of primary tumour localisation in colorectal cancer treatment and outcomes, European Journal Cancer, 84:69-80.
https://doi.org/10.1016/j.ejca.2017.07.016
Tan Z., Kan C., Sun M., Yang F., Wong M., Wang S., and Zheng H., 2022, Mapping breast cancer microenvironment through single-cell omics, Frontiers in Immunology, 13: 868813.
Wang Q., Wang Z., Zhang Z., Zhang W., Zhang M., Shen Z., Ye Y., Jiang K., and Wang S., 2020, Landscape of cell heterogeneity and evolutionary trajectory in ulcerative colitis-associated colon cancer revealed by single-cell RNA sequencing, Chinese Journal of Cancer Research, 33: 271-288.
https://doi.org/10.21147/j.issn.1000-9604.2021.02.13
PMid:34158745 PMCid:PMC8181874
Winterhoff B., Maile M., Mitra A., Sebe A., Bazzaro M., Geller M., Abrahante J., Klein M., Hellweg R., Mullany S., Beckman K., Daniel J., and Starr T., 2017, Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells, Gynecologic Oncology, 144(3): 598-606.
https://doi.org/10.1016/j.ygyno.2017.01.015
Wu F., Fan J., He Y., Xiong A., Yu J., Li Y., Zhang Y., Zhao W., Zhou F., Li W., Zhang J., Zhang X., Qiao M., Gao G., Chen S., Chen X., Li X., Hou L., Wu C., Su C., Ren S., Odenthal M., Buettner R., Fang N., and Zhou C., 2021, Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer, Nature Communications, 12(1): 2540.
https://doi.org/10.1038/s41467-021-22801-0
Xiang R., Fu J., Ge Y., Ren J., Song W., and Fu T., 2021, Identification of subtypes and a prognostic gene signature in colon cancer using cell differentiation trajectories, Frontiers in Cell and Developmental Biology, 9: 705537.
https://doi.org/10.3389/fcell.2021.705537
Yu L., Wang X., Mu Q., Tam S., Loi D., Chan A., Poon W., Ng H., Chan D., Wang J., and Wu A., 2023, scONE-seq: A single-cell multi-omics method enables simultaneous dissection of phenotype and genotype heterogeneity from frozen tumors, Science Advances, 9(1): eabp8901.
https://doi.org/10.1126/sciadv.abp8901
PMid:36598983 PMCid:PMC9812385
Zhang Q., Liu Y., Wang X., Zhang C., Hou M., and Liu Y., 2023, Integration of single-cell RNA sequencing and bulk RNA transcriptome sequencing reveals a heterogeneous immune landscape and pivotal cell subpopulations associated with colorectal cancer prognosis, Frontiers in Immunology, 14: 1184167.
https://doi.org/10.3389/fimmu.2023.1184167
PMid:37675100 PMCid:PMC10477986
Zhu J., Lian J., Xu B., Pang X., Ji S., Zhao Y., Lu H., 2023, Neoadjuvant immunotherapy for colorectal cancer: Right regimens, right patients, right directions?, Frontiers in Immunology, 14: 1120684.
https://doi.org/10.3389/fimmu.2023.1120684
PMid:36949951 PMCid:PMC10026962
(1).png)
. FPDF(win)
. FPDF(mac)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Jiahao Zhu
. Jie Lian
. Haibo Lu
Related articles
. Intratumoral heterogeneity
. Single-cell RNA sequencing
. Colorectal cancer
. Cancer stem cells
. Tumor microenvironment
Tools
. Post a comment